Newsroom
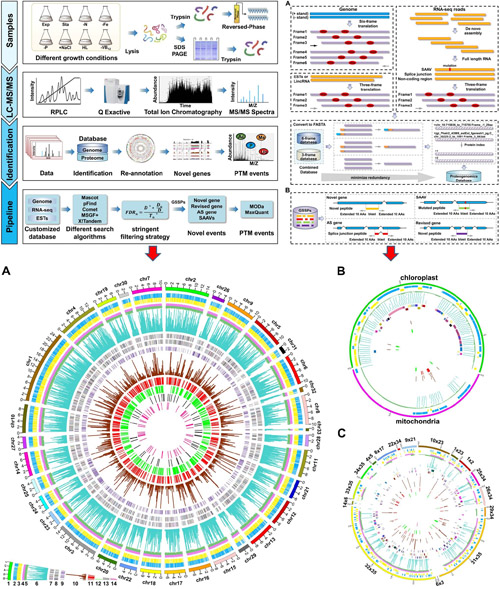
GAPP: A Proteogenomic Software for Genome Annotation and Global Profiling of Posttranslational Modifications in Prokaryotes
While the number of sequence prokaryotic genomes is growing rapidly, experimentally verified annotation of prokaryotic genome remains patchy and challenging. Recently, proteogenomics has been applied to the identification of previously unidentified genes and the correction and validation of predicted genes, as well as the identification of posttranslational modifications (PTMs) of existing gene in various organisms. Meanwhile, the computational proteomics has also become a drastically growing field, and a handful of tools have been developed to execute complete proteogenomic analyses. However, there is still a lack of automated software for proteogenomic analyses that incorporate both genome annotation and proteome-wide PTM analysis.
The research group led by Dr. GE Feng at Institute of Hydrobiology, Chinese Academy of Sciences (IHB) developed a one-stop open source software termed GAPP, which provides a complete proteogenomic pipeline for carrying out both genome annotation and large-scale PTM analysis on a proteome-wide level against prokaryotes. It is an open source and publicly available on Sourceforge. A single command is sufficient to convert data formats, create and search databases using one or more search engines, analyze and integrate results statistically, calculate false discovery rates (FDRs), group and annotate proteins, and discover and annotate PTM events globally. Thus, GAPP is accessible to a broad class of users including labs with limited bioinformatic skills and could be applied as a standard part of the genome annotation projects.
To test the effectiveness and versatility of GAPP for annotating prokaryotic genomes, researchers used GAPP to perform an in-depth proteogenomic analysis of Helicobacter pylori strain 26695 (H. pylori), one of the major human pathogens that are responsible for many gastric diseases such as duodenal ulcers and gastric cancer. Notably, about 84.9% (1,248) of the existing predicted H. pylori proteins were identified with at least two unique peptides, indicating the high coverage of protein identification. More importantly, researchers identified 20 novel protein-coding genes and modified four existing gene models in H. pylori. Likewise, a total of 1,083 proteins contained various PTM events were also detected in H. pylori.
The results were published in Molecular & Cellular Proteomics entitled “GAPP: a proteogenomic software for genome annotation and global profiling of posttranslational modifications in prokaryotes”.
This study is supported by grants from the National Key Research and Development Program (2016YFA0501304), National Basic Research Program of China (973 Program, 2012CB518700), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB14030202) and the National Natural Science Foundation of China (Grant No. 31570829).
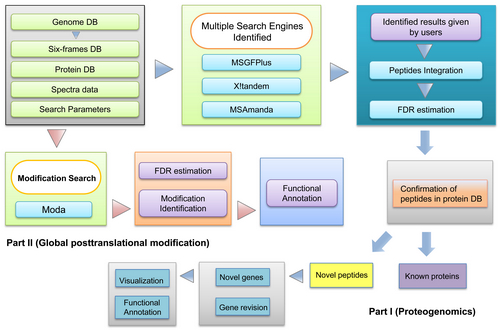
A flowchart of the GAPP workflow (Image by IHB)