Newsroom
CAS Scientists Expand Known Phosphorylation Sites in Tetrahymena from Dozens to Thousands
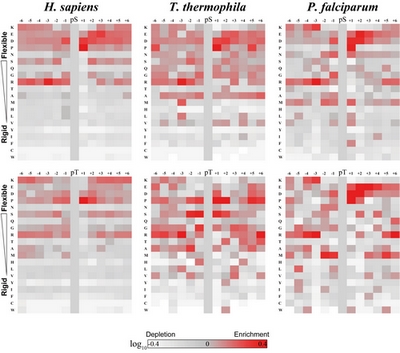
CAS researchers plotted heat maps to reveal the enrichments /depletion of amino acid residues flanking the protein phosphorylation sites in T. thermophila, H. sapiens and P. falciparum. Comparative analysis of these heat maps highlights the enrichments of flexible residues, and other conserved and species-specific features of amino acid residues flanking the phosphorylation sites in T. thermophila.(Image/IHB)
Tetrahymena thermophila is a species of free-living unicellular ciliated protozoa that is widely distributed in freshwater environments around the world. A long tradition of fundamental research on T. thermophila has contributed to numerous scientific breakthroughs such as the discovery of catalytic RNA, telomere structure and telomerase activity. Notably, 1069 protein kinases are predicted in the T. thermophila genome. Even compared with the kinome of H. sapiens containing 518 protein kinases, the kinome of T. thermophila is significantly extended. However, only a few dozen phosphorylation sites in T. thermophila are known, presenting a major obstacle for further understanding the regulatory roles of reversible phosphorylation in this organism.
Recently researchers from Institute of Hydrobiology, Chinese Academy of Sciences (IHB) and Institute of Biophysics, Chinese Academy of Sciences (IBP) reached a millstone in T. thermophila phosphoproteomic study. They conducted the first global and site-specific phosphoproteome profiling of T. thermophila by using high accuracy mass spectrometry-based proteomics techniques. In total, 1384 phosphopeptides and 2238 phosphorylation sites from 1008 T. thermophila proteins were identified through the combined use of peptide prefractionation, TiO2 enrichment, and 2D-LC-MS/MS analysis. Gene Ontology analysis and KEGG pathway mapping have shown that the identified phosphoproteins are implicated in the regulation of various biological processes such as transport, gene expression, and mRNA metabolic process.
The feature of amino acid residues flanking the phosphorylation sites in T. thermophila was also been characterized. CAS researchers found that phosphorylation motif pattern in this organism was highly affected by its unique kinome and A-T rich genome. Additionally, compared to other organisms, T. thermophila possess high PIKK kinase family recognition motif, which give the researchers clue that protein belong to this family may play a vital role in T. thermophila life cycle maintenance.
One of the most challenging problems in phosphoproteome studies is how to match the phosphoproteome to the kinome accurately. Up to now, the kinase recognition motif-based prediction is the most widely used method to predict the kinase-substrate relationships. But it is still infeasible to match a phosphorylation site to a specific protein kinase as some protein kinases may share the same or similar recognition motifs. Therefore, the researchers performed the integrated analysis of the phosphoproteomics data with TGN data to improve the accuracy of motif-based kinase-substrate predictions and infer the potential upstream protein kinases of the identified T. thermophila phosphorylated proteins.
By using the integrated analysis, the researchers have successfully identified an evolutionary conserved MAPK signaling pathway and other putative signaling pathways in this organism. Therefore, the integrated analysis of phosphoproteomics data with transcripteomics data provide a point of departure for more in-depth studies in understanding the physiological functions underlying phosphorylation and facilitate the elucidation of the entire signaling networks in the specific context of T. thermophila.
Above analysis from CAS researchers provided the first global survey of phosphorylation in T. thermophila using a phosphoproteomic approach and suggest a wide ranging regulatory scope of this modification. The results have been published online in Molecular & Cellular Proteomics entitled "Phosphoproteomic analysis of protein phosphorylation networks in Tetrahymena thermophila, a Model Single-Celled Organism"